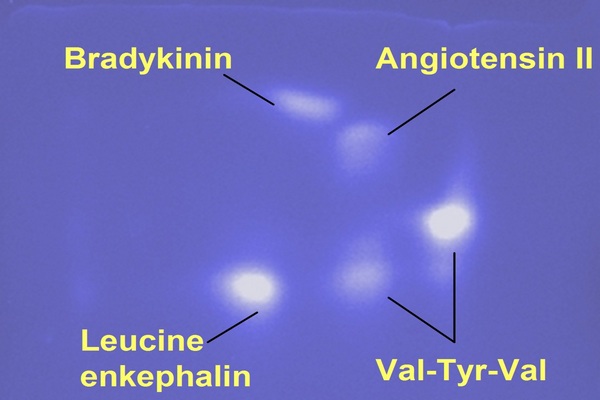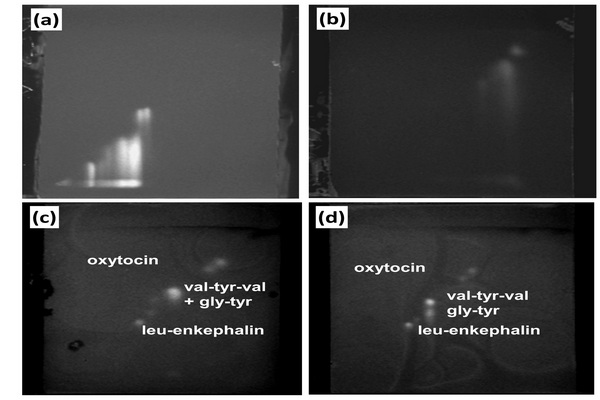
Figure 5.1
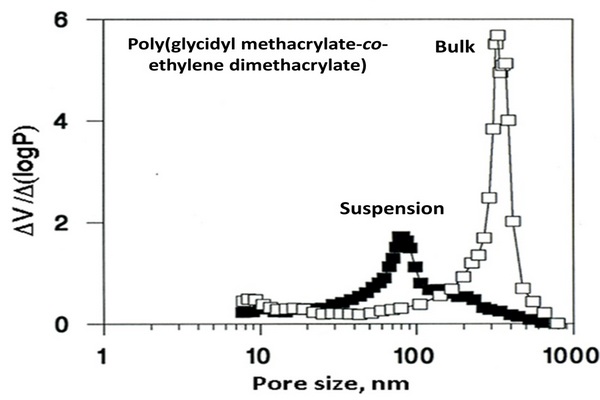
Differential pore size distribution plots of the poly(glycidyl methacrylate-co-ethylene dimethacrylate) beads and monolith prepared from the same polymerization mixture consisting of glycidyl methacrylate (24%), ethylene dimethacrylate (15%), cyclohexanol (48%), dodecanol (12%), and azo-bis-isobutyronitrile (1% with respect to monomers) at a temperature of 70oC.
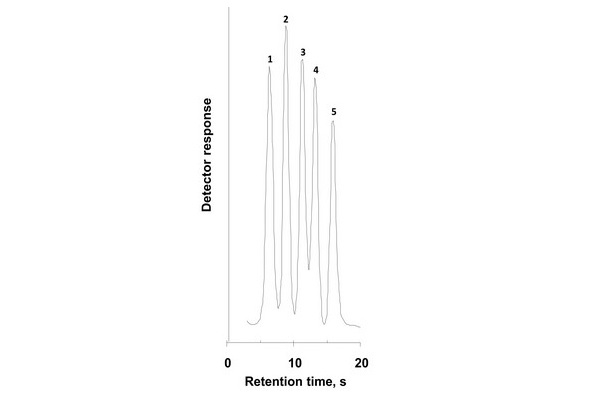
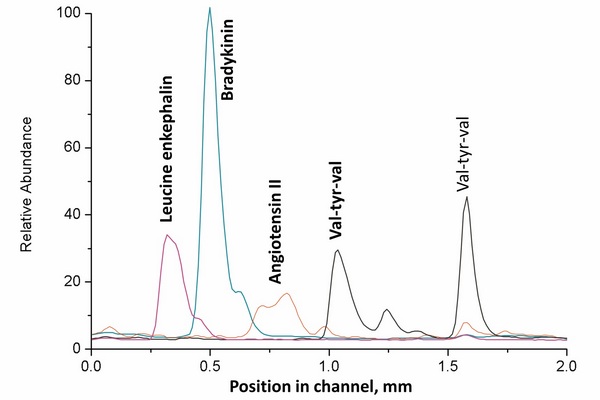
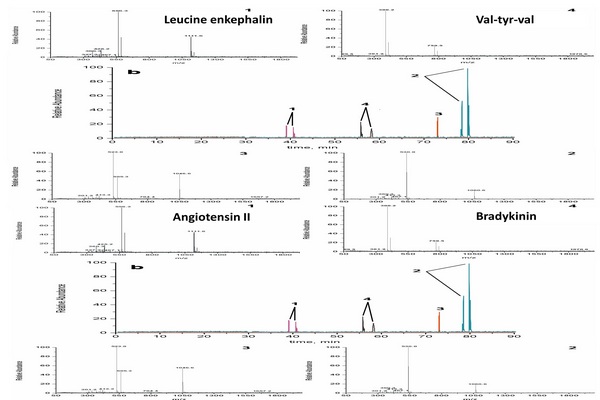
Figure 5.2
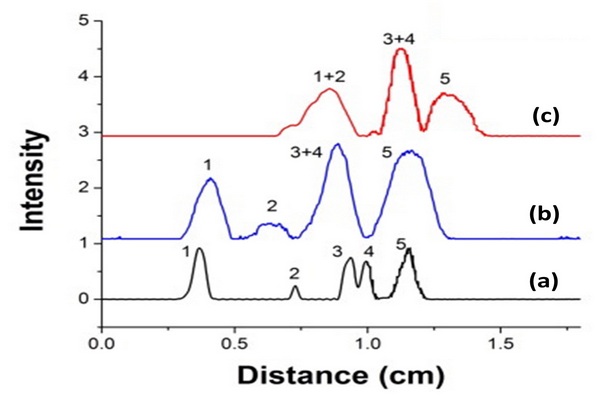
Fast reversed-phase separation of proteins. Conditions: Poly(styrene-co-divinylbenzene) monolithic column 50 x 4.6 mm I.D., mobile phase gradient 42%-90% acetonitrile in 0.15% aqueous trifluoroacetic acid in 0.35 min, flow rate 10 mL/min, UV detection at 280 nm. Peaks: ribonuclease (1), cytochrome c (2), bovine serum albumin (3), carbonic anhydrase (4), and ovalbumin (5).
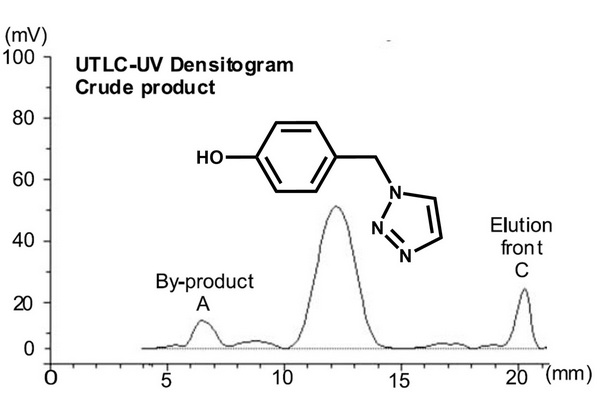
Figure 5.3
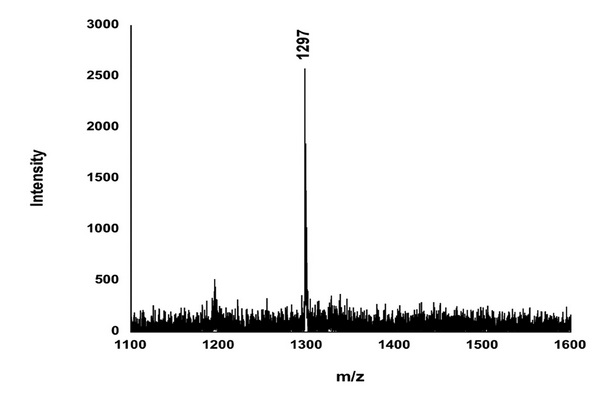
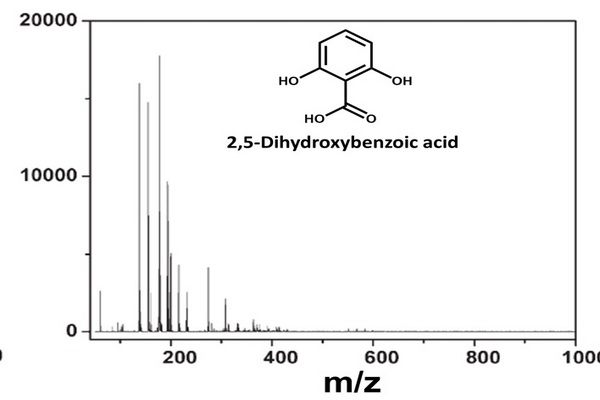
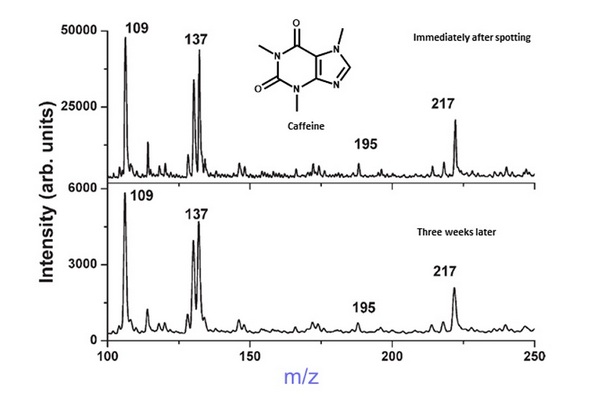
The separation of crude products from synthesis of triazole (m/z 176) using the mobile phase ethyl acetate n-hexane (1:2 v/v) with 2% acetic acid and presented as ultrathin layer chromatography (UTLC)-UV densitogram of the synthesis sample (top). Identification of peaks was achieved using atmospheric pressure matrix-assisted laser desorption/ionization (MALDI)-mass spectrometry (MS) spectra of the by-product A (m/z 369) (center) and the product (m/z 198 [M+Na]+ and m/z 107) (bottom). The main matrix ions are marked with asterisks.

Figure 5.4
Scanning electron microcope images illustrating polyacrylonitrile layer prepared “conventional” electrospinning (A) and aligned electrospun polyacrylonitrile nanofibers generated on the rotating collector at rotational speeds of 1250 rpm (B).